Coordinates in bioinformatics
0-based programs or file formats:
0-based index is often the convention in computer science
- BAM, BCFv2, BED, and PSL formats (https://samtools.github.io/hts-specs/SAMv1.pdf)
- IGV (https://software.broadinstitute.org/software/igv/IGV, “the end position
is excluded”)
- However, be noted that the display is one-based!. Please see below for comparison between IGV, UCSC genome browser display, and python string handling.
- pysam (always 0-based, following convention of Python, http://pysam.readthedocs.io/en/latest/api.html)
e.g.
ATCG
01234 <- coord
beg: 0
end: 3
seq: [0, 4) # excluding end
1-based programs or file formats:
1-based index may be more intuitive for visualization.
- SAM, VCF, GFF and Wiggle formats (https://samtools.github.io/hts-specs/SAMv1.pdf)
e.g.
ATCG
1234 <- coord
beg: 1
end: 4
seq: [1, 4] # including end (the file formats may still be excluding the end, UNCONFIRMED)
Comparison of IGV, UCSC genome browser and Python string handling
Here, we look at one particular position, chr3:38184488
IGV centers at T
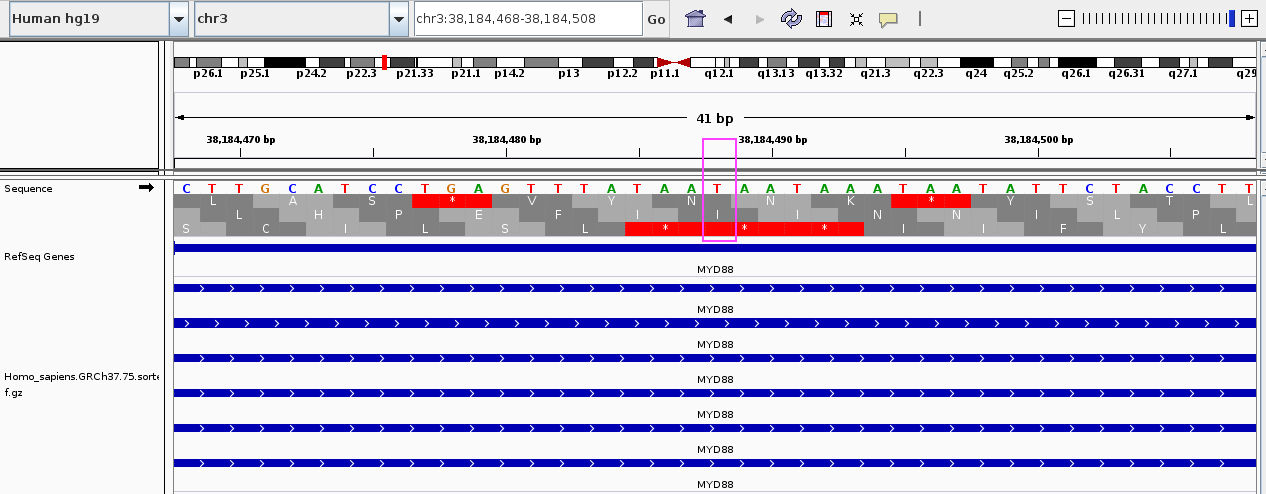
UCSC genome browser also centers at T, showing views with 3x and 10x zoom-out, respectively
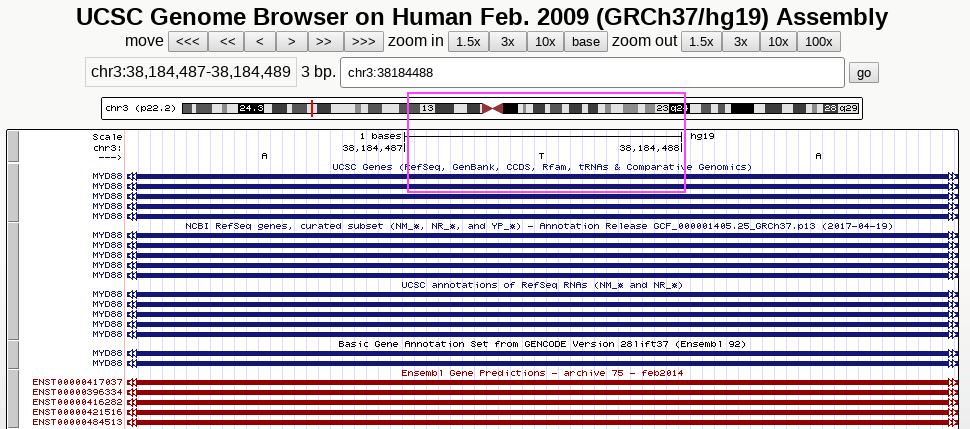
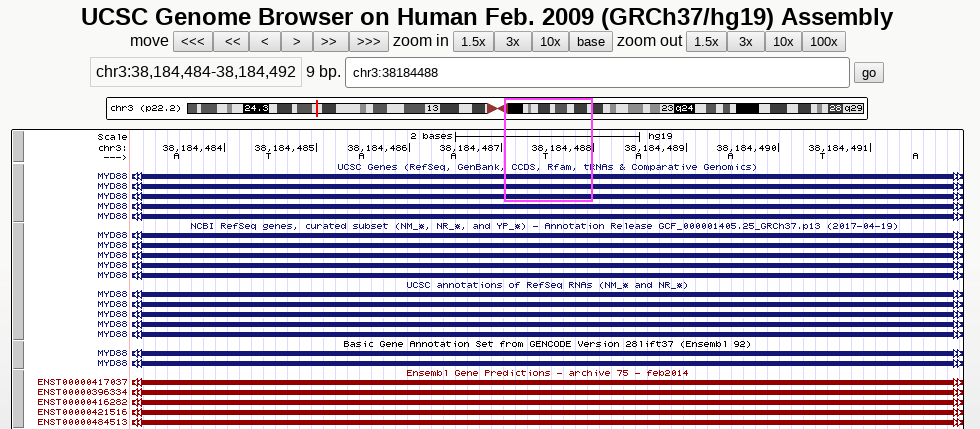
Python string handling
In [0]: import pysam
In [1]: fa = pysam.FastaFile('/gsc/www/bcgsc.ca/downloads/tasrkleat-static/on-cloud/hg19.fa')
In [2]: fa
Out[2]: <pysam.libcfaidx.FastaFile at 0x7f622023f828>
# fetch chr3 sequence
In [3]: seq = fa.fetch('chr3')
# seq is a plain python string
In [4]: type(seq)
Out[4]: str
# chr3 length
In [5]: len(seq)
Out[5]: 198022430
# python string is 0-based, so 38184488 actually points to the position 38184489
# in IGV/UCSC
In [6]: seq[38184488]
Out[6]: 'a'
# 38184487 correspond to the visualization above
In [7]: seq[38184487]
Out[7]: 't'
# verify the surroundings, python string slicing convention: excluding end, i.e. [beg, end)
In [8]: seq[38184486: 38184488 + 1]
Out[8]: 'ata'
In [9]: seq[38184485: 38184489 + 1]
Out[9]: 'aataa'
In [10]: seq[38184484: 38184490 + 1]
Out[10]: 'taataat'
In [11]: seq[38184483: 38184491 + 1]
Out[11]: 'ataataata'